3.2. Cinétique de dépôt
La vitesse de dépôt est déterminée à partir de l’épaisseur d
maximale3
de la couche et du temps de dépôt t. Les mesures d’épaisseur sont effectuées avec le
« TALYSTEP », décrit dans l’annexe A.1. En divisant d par t, on obtient la vitesse
V d. Ceci sous-entend que la vitesse de croissance est constante durant tout le dépôt,
ce qui a été vérifié par une série de dépôts de durées différentes. Un temps
d’incubation est observé seulement pour des dépôts à très basses pressions de type
UHVCVD4 [74].
3.2.1. Le choix de gaz de dilution
Durant les premiers essais avec ce nouveau
réacteur, l’hydrogène a été utilisé comme gaz de dilution injecté dans la partie
basse du réacteur. Les vitesses de dépôt se situaient autour de 6 µm/h
pour une pression totale de 100 mbar et une température de 550 ℃ du
substrat sur la plaque en inox (figure 2.2). Après l’installation de la nouvelle
plaque chauffante en graphite (figure 2.3), on a constaté une diminution
importante de la vitesse tout en gardant les mêmes paramètres. Dans le tableau
3.1
| TAB. 3.1: | Vitesse de dépôt en µm/h à 550 ℃ pour plusieurs pressions totales.
La vitesse avec la nouvelle plaque en graphite est seulement d’environ 10 %
de la valeur constatée avec la plaque métallique. |
| | 10 mbar | 25 mbar | 50 mbar | 100 mbar
| | inox & cuivre | 0,48 | 2,1 | 3,8 | 5,7 |
| graphite | 0,17 | 0,24 | 0,3 | 0,41 |
| |
|
|
sont inscrites les valeurs de la vitesse de dépôt à différentes pressions pour
une température de 550 ℃. Elles sont maintenant environ dix fois plus
faible. Ceci peut s’expliquer par le fait de la déformation de la plaque en
inox, qui non seulement a provoqué une dégradation du contact thermique
entre les disques en cuivre et la surface en inox, mais aussi un préchauffage
de l’hydrogène comme expliqué sur la page 53. De plus la conductivité
thermique du graphite est 5 à 16 fois plus élevée que celle de l’inox (voir
tableau 2.1), ce qui conduit à l’obtention d’un bloc de métal nettement plus
chaud que le bloc en graphite pour une même température de substrat. Une
vérification de la puissance injectée permet de consolider cette hypothèse :
les courants dans les résistances coaxiales sont maintenant plus faibles.
On se retrouve donc avec une température plus basse du gaz et ainsi une
moindre efficacité des réactions chimiques en phase gazeuse (équations 1.2 et
1.3).
Afin de retrouver des vitesses élevées de dépôt, l’hydrogène a été
remplacé par de l’hélium, un choix qui est fait également par d’autres
chercheurs.5
D’après les équations de réaction 1.2, 1.4 et 1.5 il est évident que la pression
partielle d’H2 entre en jeu dans la cinétique de décomposition du SiH4. Cette
influence peut être décrite par la loi classique de LANGMUIR-HINSHELWOOD
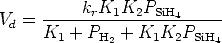 | (3.3) |
où kr, K1 et K2 sont des constantes de réaction et d’équilibre [64]. PSiH4 et PH2
sont les pressions partielles de SiH4 et H2 respectivement. Puisque H2 est un produit
de la pyrolyse de silane, une pression partielle plus élevée ralentit la décomposition
en phase gazeuse.
L’équation 3.3 n’est pas applicable dans notre cas d’une manière exacte, en
raison du grand nombre d’inconnues. Comme le mélange des gaz se fait dans le
réacteur, les pressions partielles des deux gaz peuvent être très différent
selon les endroits dans l’enceinte, de plus on ne connaît pas précisément les
gradients de température. Néanmoins elle montre, que la concentration de H2
diminue la vitesse de croissance, ce qui est aussi rapporté par d’autres
auteurs [16, 97].
L’augmentation de la vitesse par le changement de gaz diluant est illustrée dans
la figure 3.2.

| FIG. 3.2: | Vitesses de dépôt en nm/min à 550 et 580 ℃ pour plusieurs
pressions totales : avec l’hélium comme gaz diluant au lieu de H2 on augmente
la vitesse de dépôt. |
|
On peut remarquer, que la vitesse est exaltée surtout aux pressions élevées. Ceci est
probablement dû à un effet supplémentaire, liant la vitesse de dépôt avec la pression
de H2 : à des basses températures, la désorption de H2 peut devenir le facteur, qui
limite la vitesse de croissance [6, 97]. Plus la pression est haute, plus cette
désorption est empêchée.
3.2.2. Influence de la température sur la vitesse de dépôt
Afin d’étudier
l’influence de la température sur la vitesse de dépôt, on a réalisé des couches
minces à 5 températures de substrat différentes, à savoir 540, 560, 580, 600
et 620 ℃. À des températures plus élevées la probabilité de rupture des
résistances chauffantes devient trop importante, à plus basse température les
vitesses de dépôt s’approchent de celles déjà atteintes par la technique
LPCVD.
Les vitesses de dépôt V d sont tracées en échelle logarithmique dans la figure
3.3

| FIG. 3.3: | Vitesses de dépôt en nm/min à plusieurs pressions totales P en
fonction de la température du substrat. |
|
en fonction de 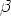 = 1/kT avec la constante de BOLTZMANN k = 8,617 × 10-5 eV K-1
et la température absolue T.
Avec des techniques CVD basées sur la décomposition thermique dans un
réacteur à paroi chaude, par exemple la LPCVD du laboratoire GMV, où la
température est homogène, la vitesse de dépôt V d suit souvent une loi
exponentielle
= 1/kT avec la constante de BOLTZMANN k = 8,617 × 10-5 eV K-1
et la température absolue T.
Avec des techniques CVD basées sur la décomposition thermique dans un
réacteur à paroi chaude, par exemple la LPCVD du laboratoire GMV, où la
température est homogène, la vitesse de dépôt V d suit souvent une loi
exponentielle
 | (3.4) |
en fonction de la température absolue T. La valeur de Ea est l’énergie d’activation
apparente et on parle d’une réaction thermiquement activée.
Comme on peut le remarquer sur la figure 3.3, cette loi n’est pas respectée dans
notre réacteur sur toute la gamme de température. Ceci peut s’expliquer par le fait,
que la température n’est pas répartie d’une façon homogène. Notamment, la
température notée T est celle du substrat ; la phase gazeuse, dont les réactions
peuvent limiter la vitesse de dépôt, est plus froide. D’ailleurs, il existe une
complexité de mécanismes qui sont intégrés avec leurs propres cinétiques dans la
croissance de la couche :
- la cinétique des réactions dans la phase gazeuse, qui est dépendante des
pressions partielles des réactants et de la température locale ;
- le transport de masse vers la surface de substrat, qui est contrôlé par les
flux de gaz et surtout les longueurs de diffusion ;
- le transport de chaleur, puisque les gaz rentrent dans la chambre à
température ambiante ;
- les réactions en surface, qui sont accélérées par une augmentation de
température ;
- la désorption des produits de réaction, dans notre cas l’hydrogène, à la
surface.
Lorsqu’un processus, qui suit la loi d’ARRHÉNIUS 3.4, détermine
principalement la vitesse de toute la chaîne des réactions dans un certain
régime6,
il est possible d’attribuer la valeur de l’énergie d’activation apparente à ce
processus.
Nos courbes de la figure 3.3 ne montrent pas de tels régimes distincts. On
constate cependant une augmentation décélérée de la vitesse de dépôt avec la
température.
3.2.3. Influence de la pression sur la vitesse de dépôt
En traçant les mêmes
valeurs V d en fonction de la pression, on obtient les courbes de la figure
3.4

| FIG. 3.4: | Vitesses de dépôt en nm/min à plusieurs températures de substrat
T en fonction de la pression totale P. |
|
pour les cinq séries de dépôt à différentes températures. On peut observer une
exaltation importante de la vitesse de dépôt avec une pression totale croissante.
Cette augmentation est d’autant plus forte, que la température est élevée et suit
assez bien la loi de LANGMUIR-HINSHELWOOD 3.3, comme c’est en général le cas
pour la LPCVD [27].
À des pressions élevées au dessus de 400 mbar, une saturation et même une
atténuation de la vitesse de croissance pour les séries à 540 et 560 ℃ sont
observées. Un tel comportement peut être également supposé pour les séries à plus
haute température. On constate par ailleurs une limitation de la pression lorsque la
température augmente. Cette limitation est imposée par le phénomène de
pulvérulence qui se manifeste aux grandes vitesses [27, 28].
3.2.4. Discussion et interprétation
La forte décélération de l’augmentation de V d
à température et pression croissante peut être expliquée en termes de régime de
diffusion et de régime de réactions. En effet, les réactions en surface et en phase
gazeuse montrent une forte dépendance de la température, ce qui ce traduit par une
forte augmentation de V d. Pour la désorption de l’hydrogène de la surface de
silicium par exemple, des énergies d’activation de 2,1 eV à 2,3 eV sont
rapportées [6, 97]. La décomposition du silane (réaction 1.2) présente également
une activation de l’ordre de 2,3 eV pour Td autour de 550 ℃ [17]. Nous observons
des énergies d’activation encore plus élevées, surtout pour des faibles températures
de substrat, ce qui peut être expliqué par la basse température de la phase
gazeuse : en effet, en dessous de 500 ℃, le silane ne se décompose quasiment
pas [42].
Pour les pressions très élevées, la vitesse de dépôt est gouvernée par le régime de
diffusion à travers l’activation thermique du coefficient de diffusion des espèces en
phase gazeuse, qui est relativement faible. La saturation de V d en fonction de la
pression confirme cette hypothèse. D’autres auteurs ont rapporté des augmentations
de V d également très faibles, pour des températures autour de 600 ℃ et des
pressions de l’ordre d’1 mbar [44, 97].